EY新日本有限責任監査法人 ライフサイエンスセクター
公認会計士 岸 佳祐/恒田 範/辻󠄀 港人
1. はじめに
近年では、新型コロナウイルス感染症に関連した治療薬の承認申請や、研究開発の断念といったニュースが報道されるほか、患者数が少なくこれまで治療法が確立されていない疾患、いわゆる「アンメット・メディカル・ニーズ」に対する治療薬の開発にも注目が集まっています。それに伴い、製薬企業の研究開発の動向や新薬の種を持つベンチャー企業の買収に対しての期待が一層高まっています。
2. 新薬創出プロセス
新薬を一つ生み出すためには、一般的に少なくとも10年の歳月と、数百億円から数千億円となる投資が必要とされています。これは「医薬品」が直接、生命にかかわるものであることから、新薬開発において安全性、有効性について念入りなチェックを行うプロセスと存在するためです。
新薬開発の工程は、第一段階では新しい化学物質を作り出し、医薬品としての利用可能性があるものを選び出すことから始まり(新薬の探索)、第二段階では「非臨床試験」と呼ばれる、動物により薬効・物性・体内動態・毒性を確かめる研究評価を行い、「医薬品の候補」を選定します。第三段階では「臨床試験」と呼ばれる、ヒトを対象とした試験を行います。臨床試験は「治験(治療試験)」とも呼ばれ、第二段階をパスした「医薬品の候補」(治験薬)を用いて、病院などの医療機関において同意を得た健康な人や患者を対象に、国の承認を得るために必要なデータを集める試験のことを言います。
これらの第一段階から第三段階までに得られた成績をまとめて厚生労働大臣に承認申請を行い、審査された後、承認に至ります。承認された医薬品は薬価基準に収載され、初めて販売されることとなります。製品化に至るまでには、かなりの医薬品がふるい落とされるため、合成化合物(新薬の候補)について、医薬品として製造・販売される確率は2万2000分の1(日本製薬工業協会調べ)といわれています。
ゆえに新薬の開発には、膨大な開発期間、巨額の開発資金が費やされ、かつ極めて高い投資リスクを製薬会社は負うこととなります。
冒頭で一般的に少なくとも10年の歳月がかかると述べましたが、希少疾患等であれば、臨床試験のための十分な人数を集めることが困難な場合があります。そのような希少疾患を対象とした医薬品の開発においては、日本では、国から指定を受けたうえで、製造販売後の調査(全例調査)等を行うことを条件に承認が行われるケースもあります。また、新型コロナウイルス感染症のワクチンや治療薬をめぐっては、その緊急性から迅速な対応が必要であると判断され、「特例承認」や「緊急承認」が行われました。新型コロナウイルス感染症のワクチンは過去からベースになる部分の研究が行われていたとはいえ、研究開発が加速度的かつ同時並行的に行われ、特例承認や緊急承認などの制度を使ったことから異例の速さで承認が行われました。「緊急承認」は、通常の臨床試験が完了していないものについても有効性が推定されれば、患者から文書による同意を得て初めて投与される必要があるといった条件付きで承認され、特例承認よりさらに迅速に承認を行うことができる制度となっています。特にワクチンでは、海外での検証的臨床試験の結果のみでも承認可能となりました。
| 通常承認 | 条件付早期承認 | 特例承認 | 緊急承認 | |
| 対象 | すべての医薬品等 | 希少疾患医薬品等 | 海外で流通している医薬品等 | すべての医薬品等 |
| 有効性 | 確認 | 確認 | 確認 | 推定 |
| 安全性 | 確認 | 確認 | 確認 | 確認 |
3. 動向
これまで国内外で製薬企業同士の合併が繰り返され、他社からある程度研究開発の進んだ新薬候補化合物の開発権を取得すること(「ライセンスイン(導入取引)」)が行われてきました。過去には、日本の製薬企業は生活習慣病の領域の低分子創薬で数々のブロックバスター(大型新薬)を輩出してきました。しかし、副作用が多いことやバイオテクノロジーの進化により、低分子創薬からバイオ医薬品・バイオ後続品(バイオシミラー)の開発にシフトしています。また、疾患領域も生活習慣病等から希少疾患等の領域へ拡大しています。
近年では、製薬企業においては新薬開発の難易度の上昇に伴い、成功確率を上げる努力や経営資源の選択と集中するため、疾患領域の絞り込みによる事業譲渡、新しい医療用医薬品候補化合物(パイプライン)を獲得するための買収等も繰り広げられています。また、研究開発における生産性向上やスピードアップのために製薬企業の研究開発拠点の集約も行われています。そして、上記のとおり、研究開発には多額な費用と期間が生じるため、専門企業へのアウトソースによる生産性の向上などが必要と判断するケースもあることから、製薬企業は自社で研究するのではなく、外部企業へ研究を委託し、その研究成果を利用することも多くなってきています。特に臨床試験では外部のCRO (Contract Research Organization、開発業務受託機関)を利用することも増えています。
4. 製薬業における研究開発活動
(1) 特許管理
a. 特許権の概要
膨大な研究開発投資に対する成果を保護するため、研究の過程での発明に対して特許出願が行われます。特許権により、権利者は発明を使って製品を独占的に製造・販売できることが可能となり、出願した発明が特許として認められれば、特許法により出願の日から20年間(特許法67条)、保護されることになります。また、「特許存続期間延長制度」を適用し(特許法67条の2)、最大で5年間の延長が認められています。新薬を開発した製薬企業はこの期間中に利益を確保しておく必要があります。
<図表 特許期間延長制度>
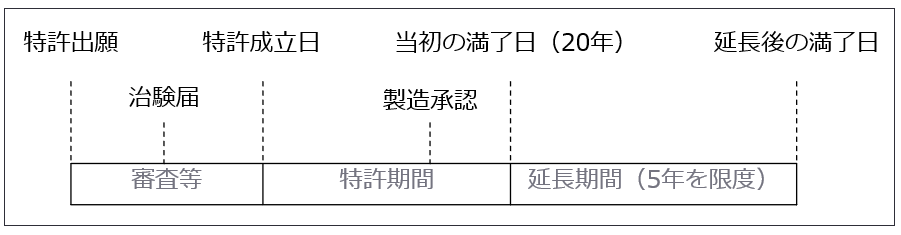
b. 特許権と後発医薬品
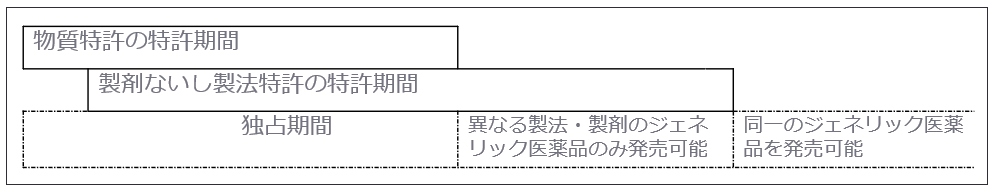
製薬企業が新薬を開発した場合、有効成分に対して物質特許が認められることが一般的ですが、通常、「新薬の特許期間の満了」とは「物質特許の期間満了」を指します。よって、ジェネリック医薬品は物質特許が満了した後に発売されることになりますが、製剤技術などの周辺特許は有効であることが多いため、ジェネリック医薬品は独自の調査・技術によって周辺特許を侵害することなく開発することが求められます。
また、物質特許自体の期間が満了しても、配合剤の特許を取得する、新たな用途特許を取得するなどにより実質的に期間を延長する場合も考えられます。
<図表 特許期間とジェネリック医薬品の関係>
c. ライセンスイン(導入取引)・ライセンスアウト(導出取引)
特許権は排他的・独占的に利用することができるものですが、通常の所有権と同様に他社に譲渡することもできれば、権利者が許諾することで他社に権利を利用させることもできます。医薬品業界においては、パイプラインを確保することが非常に重要なため、その確保を目的とした特許に係る取引も頻繁に行われるという特徴があります。
特許に係る取引はライセンス(実施許諾)契約を締結する必要があり、このような特許に係る取引は一般的に「ライセンスイン(導入取引)」・「ライセンスアウト(導出取引)」と呼ばれます。ライセンスイン(導入取引)とは製品や製造の使用許諾を獲得することで、ライセンスアウト(導出取引)とは、製品や製造の使用許諾を与えることです。
(2) 研究開発に係る会計処理
a. 研究開発費の会計処理の概要
研究開発費は、全て発生時に費用処理することとされており(「研究開発費等に係る会計基準」三、以下「研究開発費会計基準」という)、基本的な考え方として、研究開発の終了以前に発生した費用は研究開発費として処理し、終了以降に発生した費用は棚卸資産等として資産計上あるいは他の費用として処理します。この点、会計基準では製薬企業に関する研究開発の終了時点は明らかではありませんが、実務的には規制当局による製造販売承認の取得を基準として検討するものと考えられます。
b. 研究開発費の範囲
研究開発費会計基準一及び「研究開発費及びソフトウェアの会計処理に関する実務指針」第2項・第26項に基づき、製薬業における具体的な研究開発費については、例えば次のような費目が該当します。
| 研究開発のフェーズ | ||||||
| 費目 | 基礎研究 | 非臨床試験 | 臨床試験 | 承認申請と審査 | 上市後の継続調査 | |
|
||||||
c. ライセンスイン(導入取引)の処理
ライセンスイン(導入取引)の取引形態は、契約一時金支払や、一定の条件が達成されると支払われるマイルストーン・ペイメントなど様々ですが、導入時点における研究開発の段階が会計処理の検討にあたって重要な検討事項となります。この点、導入時点で規制当局による承認の可能性が高いと見込まれ、実質的に研究開発が終了していると見なせる場合には、支出金額は新薬の販売権の取得の対価として取り扱い、無形固定資産として計上することが考えられます。一方、導入時点で非臨床試験や臨床試験の段階である場合は、開発に関する成功の可能性は不確実であるものとして、支出金額を研究開発費として処理することが考えられます。
d. 委託研究開発の処理
製薬業における研究開発は、前述のとおり長期にわたることが多く、かつ多額の資金が必要とされます。製薬企業は外部企業へ研究を委託し、その研究成果を利用することも多く、特に臨床試験段階では外部のCROを利用することは一般的です。
製薬企業は委託契約に基づき一定期間ごとに研究結果の報告を受け、対価を支払うこととなりますが、契約一時金払い、期間定額払い、マイルストーン・ペイメントなど委託先の資金状況等によっても支払いパターンは様々となります。
委託研究開発については、一般的に研究の成果は委託者側に帰属するものと考えられますので、会計上、委託者側では研究開発費会計基準に沿った処理を行うこととなり、委託研究開発に係る費用は全て発生時に費用処理することとなります。
e. 研究開発費におけるIFRSと日本基準の相違点
- 自社開発の場合
IFRSでは、研究段階と開発段階の2つに区分して資産認識要件を検討し、研究段階の支出は発生時の費用として認識し、開発段階の支出は、無形資産の定義及び一般認識要件に加え、次の6つの要件全て満たすものについて資産計上することとされています(IAS第38号57項)。この点で、研究開発費を全て発生時に費用処理する我が国の基準とは異なります。
- 技術的に無形資産を完成させることのできる可能性(技術的実行可能性)
- 無形資産を完成し、使用あるいは売却する意思があること
- 無形資産を使用あるいは売却する能力があること
- 無形資産が将来の経済的便益を生成する方法を示せること
- 無形資産を開発・使用あるいは売却するための十分な技術、財務その他資源があること
- 開発中の無形資産関連支出を、信頼性をもって測定できる能力があること
製薬業における規制当局による承認は、IAS 第38号に基づく無形資産の認識要件として必ず求められるものではなく、IAS 第38号は承認前に開発費を資産化することを禁じているわけでありません。しかしながら、一般的に臨床試験段階では、規制当局による承認を取得し製品化するまでの不確定要素が大きく、将来の経済的便益の流入可能性を立証するのは困難な場合が多いといえます。医薬品の販売には規制当局からの認可が必要になるため、この認可をもって将来の経済的便益を生成する方法を示すことが可能となったと判断することが妥当であり、臨床試験の費用等、規制当局から認可を取得するまでに発生した開発費は費用として計上することが一般的と考えられます。この点、我が国の実務においても、規制当局からの承認をもって研究開発段階の終了と判断することが通常であるため、一般的な自社研究開発については、IFRSとの差異はほとんどないと考えられます
- ライセンスイン(導入取引)の処理
製薬業では、他社の仕掛中の研究開発プロジェクトを、契約一時金払いあるいはマイルストーン支払い契約により取得し、自社で開発を継続するケースが多くみられます。将来収益への不確実性があるとして費用計上するか、取得対価の存在を経済的便益流入の合理的な測定の根拠とみて資産計上するかが論点となります。これについては、取得対価にプロジェクトの成功確率が反映されている(IAS第38号第25項)ほか、取得対価は通常信頼性をもって測定可能である(IAS第38号第26項)ことから、IFRSでは将来の経済的便益の企業への流入可能性に関する認識要件を満たすものと考え、取得対価を無形資産として資産計上するのが一般的です。一方、我が国の実務においては、研究開発段階のプロジェクトの取得は原則的に研究開発費として費用計上されるので、このケースではIFRSとの差異が大きくなると考えられます。なお、取得した研究開発プロジェクトについて事後的に自社で発生する研究開発費は、自社開発にて前述したように規制当局による承認を取得し製品化するまでの不確定要素が大きく、将来の経済的便益の流入可能性を立証するのは困難な場合が多いため一般的に費用処理されることとなります。
(3) 無形資産の減損
製薬業において、無形固定資産の減損を判断するにあたっては、次のような特徴があります。以下では、特許権及び販売権について解説をしています。
a. グルーピングの単位
資産のグルーピングは、他の資産又は資産グループのキャッシュ・フローから概ね独立したキャッシュ・フローを生み出す最小の単位で行うこととされています(「固定資産の減損に係る会計基準」二6(1))。製薬業における特許権や販売権などに関しては、通常は当該権利に関連する製品ごとにキャッシュ・フローが生み出されることとなるため、製品ごとの単位で実施することになると考えられます。
b. 減損の兆候を判断するにあたっての具体的な留意点
製品の利益率は他業種に比べて高いことから、特許権や販売権に関する減損の兆候が発生する可能性は低いものの、次のような要因が発生する場合には慎重な検討が必要になると考えられます。
- 競合品の存在
より性能の高い競合品が発生した場合においては、資産又は資産グループが使用されている事業に関連して、経営環境が著しく悪化する可能性がある - 製品の欠陥や副作用が発生した場合
製品の欠陥が明らかになった場合や、副作用が発生しリコールが生じた場合においては、当該製品の販売中止に至る可能性もあり、資産又は資産グループが使用されている事業に関連して、経営環境が著しく悪化する可能性がある。
また、仕掛中の研究開発の成果に関して資産計上を行った場合においては、製品の開発状況に関して、次の検討を行うことが必要となります。
- 当初の計画と比較して製品の開発の進捗が大幅に遅れていないか。
- 開発中止に至っていないか。
- 競合品の開発が先行する、開発費が想定以上に発生するなど何らかの要因によって当初の計画自体の見直しが必要な状況になっていないか。
c. 無形資産の減損におけるIFRSと日本基準の相違点
- 耐用年数を確定できない資産は償却を行ってはならないと規定されており(IAS第38.号107項)、未だ使用可能ではない無形資産に該当する仕掛研究開発費資産と合わせて、減損の兆候の有無に関わらず減損テストを行うことが要求されます(IAS第38号108項)。実務的には、医薬品には特許期間が存在することから、耐用年数を確定できないケースはそこまで多くないと考えられるものの、企業買収において取得した被取得企業が有するブランド等については、耐用年数が確定できないと判断されることがあるため、留意する必要があります。
- 我が国の会計基準では減損損失の戻入れは禁止されていますが、IFRSには、減損の戻入れの概念があり、戻入れの兆候を毎期各報告期間の末日において検討し、該当する場合は、償却分を調整した当初の帳簿価額を超えないように新たに見積った回収可能価額を上限として、減損損失を戻し入れる必要があります。
- その他、IFRSではのれんは償却されず、こちらも減損の兆候の有無に関係なく、毎期決まった時期に減損テストを実施する必要があります。
5. 製薬業における収益獲得活動
(1) 収益認識
企業会計基準第29号「収益に関する会計基準」(以下、「収益認識会計基準」という)、及び企業会計基準適用指針第30号「収益認識に関する会計基準の適用指針」(以下、「収益認識適用指針」といい、これらを合わせて「収益認識会計基準等」という)の適用により、収益は以下の5つのステップを踏まえて認識することとなりました。
| ステップ | 要求事項 |
| ステップ1 | 顧客との契約を識別 |
| ステップ2 | 契約における履行義務を識別 |
| ステップ3 | 取引価格を算定 |
| ステップ4 | 契約における履行義務に取引価格を配分 |
| ステップ5 | 履行義務を充足した時にまたは充足するにつれて収益を認識 |
医薬品業界においても、上記の5つのステップに当てはめて、どのようにして収益を認識するのかを検討する必要がありますが、以下では特に論点となりやすい「導出契約(ライセンスアウト)」に焦点を絞って解説をしていきます。
(2) 収益認識(導出契約)
製薬企業においては、開発中の新薬の開発権や販売権の全部又は一部を他の製薬企業に供与する契約(導出契約)を締結することが多くありますが、その目的・形態は様々あです。
例えば、自社が強みを持つ疾患領域に経営資源を集中させるために、パイプラインのポートフォリオの見直しを行い、非集中領域の製品を導出する契約を締結するケースや、開発権の一部を供与し、契約の相手方である提携企業と共同開発や共同販売を行うことで、自社の新薬開発におけるリスクや将来得られる便益を提携企業と共有するケースも考えられます。
このように、導出契約においては、様々な目的・形態に応じて契約が締結されるため、契約の内容・実態に応じて、導出元である企業が収益認識基準等に照らして慎重に判断していくことが必要となります。
(3) 収益認識会計基準等の適用範囲
収益認識会計基準等では、上記の5つのステップの当てはめの前に、契約相手である提携企業が「顧客」に該当するかどうかを評価することが会計処理の検討にあたっての最初の検討事項となり、「顧客」に該当しない場合には収益認識会計基準等の適用対象外となります。
ここで、収益認識会計基準第6項では、「顧客」とは、対価と交換に企業の通常の営業活動により生じたアウトプットである財又はサービスを得るために当該企業と契約した当事者を言います。契約先企業が顧客の場合は、通常、財又はサービスの支配が契約先企業に移転することになりますが、導出契約においてはその点が明確でないケースがあるため、留意が必要となります。収益認識会計基準第111項では、顧客ではないものとして「企業の通常の営業活動により生じたアウトプットである財又はサービスを獲得するためではなく、リスクと便益を契約当事者で共有する活動又はプロセス(提携契約に基づく共同研究開発等)に参加するために企業と契約を締結する当該契約の相手方」を例示しており、例えば、契約先企業が企業の通常のアウトプットを獲得するためではなく、先述した開発権の一部を供与して、共同開発や共同販売を行うケースのように、活動又はプロセスの結果としてもたらされる「リスクと便益を共有」するために契約を締結するケースもあります。
(4) ステップ2:履行義務の識別、ステップ5:履行義務の充足
導出契約においては、提携企業への様々な契約上の約束(履行義務)が含まれますが、契約の主要な構成要素として、ライセンスの供与があげられます。収益認識適用指針第61項では、ライセンスとは「企業の知的財産に対する顧客の権利を定めるもの」とされています。
履行義務の識別の検討においては、契約がライセンスを供与する約束のほかに、例えば臨床試験への参画や原薬・製品を供給する製造サービス等、他の約束を含むかどうかを整理した上で、ライセンスを供与する約束が顧客との契約における他の財又はサービスを移転する契約と別個の約束であるか否かを判断することが必要となります。
なお、収益認識会計基準第34項では、以下のいずれも満たす場合に別個のものと判定されます。
(1) 当該財又はサービスから単独で顧客が便益を享受することができること、あるいは、当該又はサービスと顧客が容易に利用できる他の資源と組み合わせて顧客が便益を享受することができること
(2) 当該財又はサービスを顧客に移転する約束が、契約に含まれる他の約束と区分して識別できること
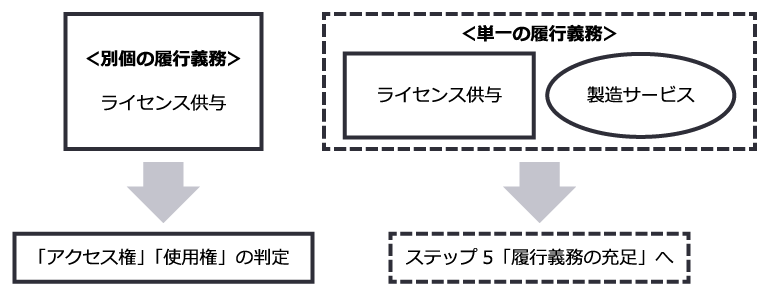
ライセンスを供与する約束が、上記に照らして、顧客との契約における他の財又はサービスを移転する約束と別個のものでない場合には、両方の約束を一括して単一の履行義務として処理し、履行義務を充足した時にまたは充足するにつれて収益を認識することとなります。
一方で、収益認識適用指針第62項によれば、ライセンスを供与する約束が、顧客との契約における他の財又はサービスを移転する約束と別個のものであり、当該約束が別個の財又はサービスと判断される場合には、ライセンスを顧客に供与する際の企業の約束の性質が、「アクセス権」「使用権」のいずれかを提供するものかを判定することになります。
| 判定 | 定義 | 収益認識の時点 |
| アクセス権 | ライセンス期間にわたり存在する企業の知的財産にアクセスする権利 | 一定の期間にわたり認識 |
| 使用権 | ライセンスが供与される時点で存在する企業の知的財産を使用する権利 | 一時点で認識 |
収益認識適用指針第63項によれば、以下の要件全てに該当する場合には、顧客が権利を有している知的財産の形態、機能性又は価値が継続的に変化しており、ライセンスは「アクセス権」を提供する約束になるとされています。
(1) ライセンスにより「顧客が権利を有している知的財産に著しく影響を与える」活動を企業が行うことが、契約により定められている又は顧客により合理的に期待されていること
(2) 顧客が権利を有している知的財産に著しく影響を与える企業の活動により、顧客が直接的に影響を受けること
(3) 顧客が権利を有している知的財産に著しく影響を与える企業の活動の結果として、企業の活動が生じたとしても、財又はサービスが顧客に移転しないこと
また、収益認識適用指針第65項によれば、以下のいずれかに該当する場合には、収益認識適用指針第63項(1)に定める「顧客が権利を有している知的財産に著しく影響を与える」ものとされます。
(1) 当該企業の活動が、知的財産の形態又は機能性を著しく変化させると見込まれること
(2) 顧客が知的財産からの便益を享受する能力が、当該企業の活動により得られること又は当該企業の活動依存していること
さらに、収益認識適用指針第150項によれば、顧客が権利を有している知的財産が「重要な独立した機能性」を有する場合には、当該知的財産の便益の実質的な部分が当該機能性から得られるため、顧客が知的財産からの便益を享受する能力は、企業の活動が知的財産の形態又は機能性を著しく変化させない限り、企業の活動による著しい影響は受けないとされています。
「重要な独立した機能性」とは、例えば「薬品の製法」も該当するものと考えられ、この場合にはライセンスが「使用権」として判定され、履行義務を充足した時に収益を認識することとなります。
参考文献・参考ウェブページ
「Q&A 業種別会計実務・4 製薬(第2版)」出版:2020年12月、著者:有限責任監査法人トーマツ
「企業への影響からみる 収益認識基準 実務対応Q&A(新版)」出版:2021年3月、著者:EY新日本有限責任監査法人
日本製薬工業協会「イノベーション創出力向上へ 産業自らの変革 (ニューズレター 2023年5月号 No.215 )」、jpma.or.jp(2024年2月19日アクセス)
ライフサイエンス
- 第1回:ライフサイエンス業界の概要と動向(上) (2024.03.05)
- 第2回:ライフサイエンス業界の概要と動向(下) (2024.03.06)
- 第3回:新薬を中心とした医療用医薬品製造販売業の概要と会計処理の特徴 (2024.03.08)
- 第4回:一般用医薬品製造販売業の概要と会計処理の特徴 (2024.03.11)
- 第5回:ジェネリック医薬品(後発医薬品)を中心とした医療用医薬品製造販売業の概要と会計処理の特徴 (2024.03.13)
- 第6回:医薬品製造受託機関(CMO)・医薬品開発製造受託機関(CDMO)の概要と会計処理の特徴 (2024.03.14)
- 第7回:医療機器製造販売業の概要と会計処理の特徴 (2024.03.15)
- 第8回:医薬品卸売業の概要と会計処理の特徴 (2024.03.18)
- 第9回:バイオベンチャー企業のビジネスと財務諸表の特徴 (2024.03.25)
- 第10回:バイオベンチャー企業におけるライセンス契約の会計処理論点 (2024.03.26)
- 第11回:ライフサイエンス関連企業における不正・コンプライアンスリスクと最近の動向 (2024.03.27)
- 第12回:バイオベンチャー企業の株式上場における論点 (2024.03.28)
- 第13回:ライフサイエンス関連企業のサステナビリティ情報開示 (2024.04.02)



EY Japan Assurance Hub
時代とともに進化する財務・経理に携わり、財務情報のみならず、非財務情報も統合し、企業の持続的成長のかじ取りに貢献するバリュークリエーターの皆さまにお届けする情報ページ